
Summary
Signal amplification methods were initially designed as an alternative to target amplification technologies, such as polymerase chain reaction (PCR), to minimize the possibility of contamination by target amplification products. Unlike target amplification, signal amplification methods (as defined herein) do not rely on enzymes for amplification. Probe-based amplification techniques, such as cleavage-based amplification and rolling circle amplification, are enzyme-based and will not be covered.
Signal amplification enhances or amplifies the signal generated from the probe molecule hybridized to the target nucleic acid sequence. Advantages of signal amplification methods include specific detection, dynamic range, ease of use, and reproducibility. To date, these methods have been challenged by advanced or automated target amplification methods such as real-time PCR. Signal amplification technologies include hybrid capture (HC) and branched DNA (bDNA) assays.
The HC method was initially developed and marketed by Digene Corporation (Gaithersburg, MD) which was acquired by Qiagen (Valencia, CA) in 2007. The bDNA method was initially developed by Chiron (Emeryville, CA), marketed by Bayer Diagnostics (Emeryville, CA). CA) CA), whose diagnostic division was acquired by Siemens (Tarrytown, NY) in 2006. Due to the increasing demand for fast detection times, automation, and multiplexing, the popularity of commercial signal amplification methods has declined in recent years. clinical virology laboratories.
Materials
The ATP CLS II bioluminescence assay kit and alkaline phosphatase were purchased from Boehringer Mannheim (Indianapolis, IN). ATP sulfurylase, adenosine 5′-phosphosulfate (APS) and 4,5′,8-trimethylpsoralen (Trioxalene) were purchased from Sigma Chemical Co., (St Louis, MO). T7 DNA polymerase was from New England Biolabs (Beverly, MA). All DNA oligonucleotides synthesized using standard cyanoethyl phosphoramidite chemistry and purified by high pressure liquid chromatography were purchased from Operon Technologies Inc. (Alameda, CA). 4-, 6-, and 8-layer DNA dendrimers and their derivatives were purchased from Genisphere Inc. (Philadelphia, PA).
Signal Amplification Cassettes (SAC)
Equimolar concentrations of the two oligonucleotide chains with complementary regions that form the two-way SAC were heated to 80 °C in a buffer consisting of 10 mM Tris-HCl, pH 8.0, 50 mM KCl, 2.5 MgCl2. mM for 3 min and slowly cooled. at room temperature to facilitate annealing. A single-stranded oligonucleotide with the ability to form a hairpin structure to generate the single-path SAC was treated in the same manner. After annealing the oligonucleotide chains to the SAC constructs, they were photocrosslinked with trioxsalen.
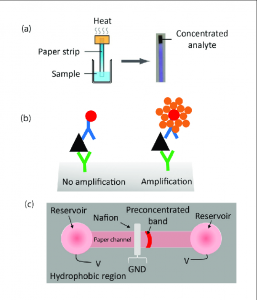
Briefly, a saturated solution of Trioxsalen in ethanol was added to a 100 µg/ml DNA solution at 1:15 (v/v) (Trioxsalen: DNA) and incubated for 30 min at 37°C. Photo-crosslinking was carried out in a Stratalinker UV Crosslinker 2400 equipped with a 365 nm light source for 45 min at 1 mJ/min on ice. These conditions, which produce 70% to 80% photocrosslinked products, were chosen by monitoring the degree of crosslinking of the radiolabeled oligonucleotides on denaturing polyacrylamide gels. After photocrosslinking, unreacted trioxsalen was removed by chloroform extraction and the SAC constructs were recovered by ethanol precipitation.
DNA dendrimers
DNA dendrimers with 4 layers (mol. weight 1.2 × 107 g/mol), 6 layers (mol. weight 1.2 × 108 g/mol), and 8 layers (mol. mol) were prepared at Genisphere Inc. Briefly, DNA dendrimers were assembled from seven oligonucleotides, strands 1–7. The seven strands were annealed pairwise in TNE200 buffer (50 mM Tris, pH 8.0, 10 mM EDTA, 200 mM NaCl) to form five basic “monomers”: A, B′, B′′, C′, and C′. ′. Chain 1 and chain 2 are hybridized to form monomer A. Similarly, chain 3 hybridized to chain 4 and chain 4 hybridized to chain 5 to produce monomers B’ and B”, respectively. . The C’ monomer was prepared by hybridizing strand 2 and strand 6 and the C” monomer was prepared by hybridizing strand 2 and strand 7. Each monomer is composed of four 31 nt single-stranded arms and a 50-base central waist.
From these building blocks, the central dendritic structure was assembled. In each assembly layer, the dendritic structure is photocrosslinked with trioxsalen. SACs were introduced by hybridization followed by photocrosslinking of an oligonucleotide that is complementary to the 3′ arms of the DNA dendrimers. This oligonucleotide provided a 30 nt long adenosine template for T7 DNA polymerase. After complete assembly, the DNA dendrimers were finally purified by ultracentrifugation in 10-50% sucrose gradients containing 50% deionized formamide at 40°C. Gradient fractions containing DNA dendrimers were pooled, ethanol precipitated, and resuspended in a buffer consisting of 50 mM Tris-HCl, pH 8.0, and 10 mM EDTA.
The nucleotide extension and cleavage-coupled signal amplification (NEESA) reaction
Signal amplification reactions were performed in 96-well U-bottom microtiter plates (solid white) from Corning Costar (Cambridge, MA) in duplicate. SACs in various configurations (monomeric, dimeric, and multimeric on DNA dendrimers) were incubated at 37 °C for 1 h in 20 µl of reaction buffer consisting of 20 mM Tris-acetate, pH 7.9, Mg(OAc)2 10 mM, 50 mM KOAc, 1 mM dithiothreitol, 250 mM TTP, 2.5 µg bovine serum albumin, 1 U T7 DNAP, 50 pmol APS, and 125 mU ATP sulfurylase. The amount of ATP generated in each reaction was measured with the ATP Bioluminescence Assay Kit CLS II from Boehringer Mannheim according to the manufacturer’s instructions.
Briefly, 45 µl of luciferase reagent was added to each well and incubated for 10 min in the dark. Bioluminescence intensity was measured using an EGG Berthold LB 96P luminometer. The mean bioluminescence value obtained from duplicate reactions was used for further analysis. In uncoupled assays, PPi was generated in the same buffer, but in the absence of APS and ATP sulfurylase, for 1 hour at 37°C. Subsequently, APS and ATP sulfurylase were added to the reaction and incubation was carried out for an additional 30 min to convert PPi to ATP.
Alkaline phosphatase-catalyzed signal amplification
Varying concentrations of alkaline phosphatase (molecular weight 140,000 g/mol) in 10 µl of 100 mM diethanolamine buffer, pH 10, were added to a 100 µl solution consisting of 100 mM diethanolamine, pH 10, 10% (v/ v) Sapphire Enhancer Solution (Tropix, Bedford, MA), 17 µl/ml CSPD (a 1,2-dioxetane substrate from Tropix), 1 mM MgCl2 and 0.02% sodium azide. After 20 min incubation in the dark, the intensity of the chemiluminescence signal was measured using an EGG Berthold LB 96P luminometer.